1. ผู้ป่วยเริ่มมีอาการทางระบบประสาท เช่น ซึมลง ชัก ในช่วงที่มารดาเริ่มงดนมผู้ป่วย มื้อกลางคืน และการตรวจทางห้องปฏิบัติการเบื้องต้น พบ nonketotic hypoglycemia แพทย์ต้องวินิจฉัยแยกโรคในกลุ่ม fatty acid oxidation defects เช่น medium chain acyl CoA dehydrogenase (MCAD) deficiency ผู้ป่วยจะเริ่มมีอาการเมื่อไม่ได้รับอาหารจากภายนอก เป็นเวลานาน หลังจากใช้ glycogen หมด ร่างกายจำเป็นต้องได้พลังงานจากการสลายไขมัน ผู้ป่วยกลุ่มนี้ไม่สามารถสลายไขมันได้ตามปกติ จึงเกิดอาการขึ้น การวินิจฉัยแน่ชัดอาจได้จากการ วิเคราะห์ organic acid ในปัสสาวะ ซึ่งจะพบ dicarboxylic acids เช่น adipic acid, suberic acid, sebacic acid การรักษาทำได้ง่าย โดยการให้อาหารบ่อยๆ ซึ่งได้ผลดีมาก
2. ผู้ป่วยที่มีพัฒนาการช้า โดยไม่สามารถอธิบายได้จากประวัติก่อนคลอด ระหว่างคลอด หรือหลังคลอด การตรวจร่างกายถ้าพบ hypopigmentation ควรนึกถึง phenylketonuria ถ้าพบ marfanoid habitus ควรนึกถึง homocystinuria การตรวจทางห้องปฏิบัติการเบื้องต้น ที่เป็นประโยชน์ คือ urine FeCl3 test ซึ่งให้สีน้ำเงินเขียวใน phenylketonuria และ Cyanide-Nitroprusside ให้สีม่วงใน homocystinuria การวินิจฉัยที่แน่นอนของทั้ง 2 โรค ได้จากการตรวจ plasma amino acid analysis การรักษาผู้ป่วย phenylketonuria ทำได้โดยการ ให้ phenylalanine ในปริมาณต่ำ ส่วนผู้ป่วย homocystinuria ครึ่งหนึ่ง จะรักษาได้ด้วยการให้ vitamin B6 เพียงอย่างเดียว ในกรณีผู้ป่วยพัฒนาการช้าหาสาเหตุไม่ได้ แพทย์ควรส่งตรวจ thyroid function test ด้วย เนื่องจากเป็นโรคที่รักษาได้ง่าย และได้ผลดี ในกรณีเป็นชาย ควรส่งตรวจ fragile X syndrome ด้วย
3. ผู้ป่วยที่มาด้วยอาการชัก ภายในอายุ 2-3 เดือนแรก ไม่สามารถอธิบายได้จากประวัติ ก่อนคลอด ระหว่างคลอด หรือหลังคลอด, การตรวจร่างกายไม่พบ dysmorphic features ที่เข้าได้กับ syndromes ใด, การตรวจทางห้องปฏิบัติการเบื้องต้น เช่น ระดับน้ำตาล ภาวะ กรดด่างในเลือด ไม่พบสิ่งผิดปกติ, CT หรือ MRI อาจปกติ หรือพบ agenesis of corpus callosum, EEG พบ burst suppression pattern, และผู้ป่วยดื้อต่อการรักษาด้วยยากันชัก แพทย์ควรคิดถึงโรค nonketotic hyperglycinemia การวินิจฉัยทำได้โดยการตรวจ plasma และ CSF amino acid analysis การรักษาโรคนี้ยังไม่ได้ผลเป็นที่น่าพอใจ
4. ในผู้ป่วยที่ระดับความรู้สึกตัวผิดปกติ มีระดับ transaminase และ ammonia ที่สูงขึ้น (Reye syndrome) ที่เป็นซ้ำ, เป็นในวัยทารก, หรือมีประวัติคนอื่นในครอบครัวเป็นด้วย ควรได้รับการตรวจเพิ่มเติมทาง metabolic รวมทั้งการวิเคราะห์ urine organic acid และ plasma amino acid
5. ในเด็กที่ปกติเมื่อแรกคลอด และมีพัฒนาการตามปกติไประยะหนึ่ง แล้วจึงเริ่มมีการถดถอย ของพัฒนาการ สูญเสียสิ่งที่เคยทำได้ การตรวจร่างกายพบ coarse facies กล่าวคือ ผมดก คิ้วดก จมูกและริมฝีปากหนา แพทย์จะต้องวินิจฉัยแยกโรคในกลุ่ม lysosomal storage disorders กลุ่มที่พบบ่อยคือ mucopolysaccharidoses ซึ่งเกิดจากการขาด lysosomal enzymes ที่ทำหน้าที่ย่อยสลาย mucopolysaccharide สารนี้จะสะสมมากขึ้นตามกาลเวลา อาการจึงเป็นมากขึ้นเมื่ออายุมากขึ้น การส่ง X-ray เป็นการตรวจกรองที่ดีมาก X-ray ที่ควรส่งคือ กะโหลก, ทรวงอก, เชิงกราน, กระดูกสันหลัง, มือและเท้า ลักษณะเฉพาะของ X-ray ในผู้ป่วยกลุ่มนี้ เรียกว่า multiplex dysostosis คือพบ J-shaped sella turcica, กระดูกซี่โครงกว้างขึ้นในส่วนปลาย ทำให้มีลักษณะคล้ายใบพาย, กระดูกสันหลังส่วนหน้ายุบตัวลง ทำให้หลังหักงอเป็นมุม, metacarpals ส่วนต้น เรียวเล็กลงเป็นตะขอ ในขณะที่ส่วนปลายกว้างขึ้น, proximal and middle phalanges หนาขึ้น และมีลักษณะคล้ายกระสุนปืน การตรวจ urine metabolic screen ด้วยวิธี cetyltrimethylammonium bromide test จะให้ตะกอนขุ่น การวินิจฉัยที่แน่ชัด ได้จากการตรวจ lysosomal enzymatic activity ซึ่งอาศัยเครื่องมือ spectrofluorometer การรักษาด้วยการปลูกถ่ายไขกระดูก อาจมีประโยชน์ในผู้ป่วยบางราย นอกจากนี้ยังมีโรค inherited metabolic disorders อีกหลายโรค ที่อาการแสดงคือการถดถอยของพัฒนาการ เช่น โรค metachromatic leukodystrophy (ภาพตัวอย่างผู้ป่วย)

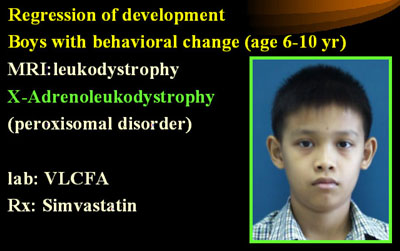
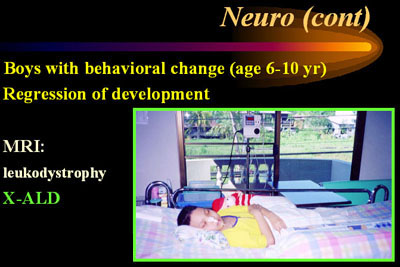
7. เด็กชายที่เริ่มมีพฤติกรรมเปลี่ยนแปลงในช่วงประถมต้น และต่อมามีการถดถอยของ พัฒนาการ เช่น เดินแล้วล้มบ่อย พูดลำบาก การมองเห็นและการได้ยินเสื่อมลง อาจได้ประวัติ ญาติผู้ชายทางฝ่ายแม่มีอาการคล้ายคลึงกัน การตรวจร่างกายอาจพบ hyperreflexia, MRI พบ leukodystrophy แพทย์ควรนึกถึงโรค X-linked adrenoleukodystrophy
 |
 |